
Current Projects
Relevant Subgraph Extraction
 One useful visualization of the large amount of data concerning genes and their relationships is a graph, also known as an interaction network. In this representation, each node in the graph is a gene and an edge connecting two nodes indicates a particular relationship between the corresponding genes. Edges can have meanings, such as indicating that one gene activates another gene or indicating that two genes belong to a common biological process. Edges can also have weights, which can represent the certainty of experimental evidence supporting the relationship or the level of sequence similarity of the gene pair, etc. For a given study, the collection of all genes and relationships creates a very dense and complex graph, even though only a subset of edges and nodes may be relevant for that study. The goal of our research is to create computational methods to extract only the subset of the graph that is strongly supported by relationships observed among genes highlighted by a given investigation.
One useful visualization of the large amount of data concerning genes and their relationships is a graph, also known as an interaction network. In this representation, each node in the graph is a gene and an edge connecting two nodes indicates a particular relationship between the corresponding genes. Edges can have meanings, such as indicating that one gene activates another gene or indicating that two genes belong to a common biological process. Edges can also have weights, which can represent the certainty of experimental evidence supporting the relationship or the level of sequence similarity of the gene pair, etc. For a given study, the collection of all genes and relationships creates a very dense and complex graph, even though only a subset of edges and nodes may be relevant for that study. The goal of our research is to create computational methods to extract only the subset of the graph that is strongly supported by relationships observed among genes highlighted by a given investigation.
Multi-Loci Prioritization
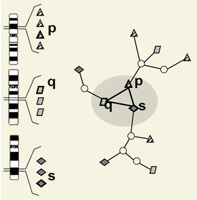 In collaboration with Prof. Yves Moreau's group at KULeuven http://www.kuleuven.be/bioinformatics/, we are developing techniques to make sense of multiple lists of genes simultaneously. Often studies produce distinct lists of genes that may contribute to the process of interest but the exact contributor from each list is not known. For example, a patient cohort may include 10 patients yet there is a different chromosomal abnormality identified in each patient. The goal is to find the gene(s) in each patient located at the site of the chromosomal abnormality in that patient that might be contributing to the disease. Our work uses a graph representation of functional relationships among genes to identify the set of genes, one or more from each patient, that closely interact with causative genes in the aberrant loci of the other patients. Another example is to find among lists of personal genetic variants for each patient a subset that might affect genes sharing a common pathway. In general, this approach is applicable whenever genes (or other genomic entities) must be prioritized within a list, yet the prioritization must be simultaneously sensitive to the prioritization of genes in other lists.
In collaboration with Prof. Yves Moreau's group at KULeuven http://www.kuleuven.be/bioinformatics/, we are developing techniques to make sense of multiple lists of genes simultaneously. Often studies produce distinct lists of genes that may contribute to the process of interest but the exact contributor from each list is not known. For example, a patient cohort may include 10 patients yet there is a different chromosomal abnormality identified in each patient. The goal is to find the gene(s) in each patient located at the site of the chromosomal abnormality in that patient that might be contributing to the disease. Our work uses a graph representation of functional relationships among genes to identify the set of genes, one or more from each patient, that closely interact with causative genes in the aberrant loci of the other patients. Another example is to find among lists of personal genetic variants for each patient a subset that might affect genes sharing a common pathway. In general, this approach is applicable whenever genes (or other genomic entities) must be prioritized within a list, yet the prioritization must be simultaneously sensitive to the prioritization of genes in other lists.
Heterogenous Data Integration
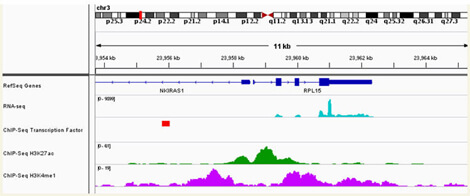
The ability to view genomic material from many different perspectives has left us with the incredible opportunity, as well as the incredible challenge, of integrating over the various interpretations. A number of our projects integrate multiple data sources, either annotation sources or primary data sources. Often we want to collect any information regarding relationships between two genes, regardless of the type of relationship and possibly inferring additional information across homologous genes in other species. In other cases, combining multiple primary data sets from distinct sources provides a more comprehensive view of the potential biological phenomenon at work. For example, viewing sequencing data of the active genes (RNA-seq), together with data from a potential regulator (ChIP-seq) and epigenetic information in the form of histone modifications (ChIP-seq) allows us to determine or predict that activity of genes and causes for their regulation.
Other Collaborative Projects
Transcriptional Dysregulation in Innate Immunity Response (with Scott Alper, PhD)
Developing a histone signature of T cell Memory (with Brian O'Connor, PhD and Ross M. Kedl, PhD)
Asthma, Histone Biology, and T cell skewing (with Brian O'Connor, PhD and Erwin W. Gelfand, MD)
Transcriptomics of Asthma (with Rafeul Alam, MD, PhD)
Kinase Transcriptomics (with Jeffrey Kern, MD and James Finigan, MD)
Mechanisms promoting translocations and mature B cell lymphomas (with Jing Wang, PhD)
Metabolic profiles of COPD phenotypes (with Russ Bowler, MD, PhD; Nichole Reisdorph, PhD; and Katerina Kechrics, PhD)
Study of populations of aveolar macrophages (with Bill Janssen, MD and Donna Bratton, MD).
Epigenetics of Sarcoidosis (with Nabeel Hamzeh, MD; Lisa Maier, PhD; and Brian O'Connor, PhD)