
Familial Pulmonary Fibrosis: Forms
This information was reviewed and approved by Jeff Swigris, DO, MS (9/1/2015).
Pulmonary fibrosis is ongoing scarring of the lungs that makes it increasingly more difficult to breathe. "Fibrosis" is the medical term for scarring.
Idiopathic Interstitial Pneumonia (IIP) is a form of pulmonary fibrosis and a subgroup of Interstitial Lung Disease (ILD).

IIP is a subgroup of interstitial lung disease. IPF is the most common form of IIP.
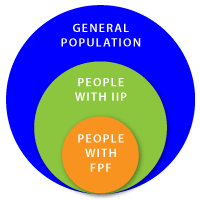
There are many different forms of IIPs (see the picture above). The different forms of IIPs are as follows:
- Idiopathic Pulmonary Fibrosis (IPF)
- Non-Specific Interstitial Pneumonitis (NSIP)
- Cryptogenic Organizing Pneumonia (COP)
- Lymphocytic Interstitial Pneumonitis (LIP)
- Respiratory Bronchiolitis associated Interstitial Lung Disease (RB-ILD)
- Desquamative Interstitial Pneumonitis (DIP)
- Acute Interstitial Pneumonia (AIP) also called Hamman-Rich Syndrome
Idiopathic Pulmonary Fibrosis (IPF) is the most common form of IIP. Every year approximately 48,000 individuals are diagnosed with IPF In the United States. Anyone can develop IPF, and the average age of onset is between the 5th and 6th decades of life.
In Idiopathic Interstitial Pneumonia the tissue in the lungs becomes inflamed and/or scarred. The interstitium is a specific area of the lung that includes the area in and around the small blood vessels and alveoli (air sacs). This is where the oxygen breathed in is exchanged with the carbon dioxide being breathed out. Inflammation and scarring of the interstitium impairs the lungs ability to extract oxygen from the air.
In the context of Idiopathic Interstitial Pneumonia the word pneumonia is being used in a very broad sense. "Pneumonia" literally means inflammation of the lung. Most commonly, the word pneumonia is used to describe a bacterial infection of the lung, but this is not the case in IIP. Infections cause inflammation - thus "pneumonia". There is no infection in Idiopathic Interstitial Pneumonia and the inflammation of the lung is occurring due to other reasons. Often the reason for the inflammation is unknown.
Any of these forms of IIP can cluster in families. It is estimated that about 10-15% of people with an IPF have the familial form, Familial Pulmonary Fibrosis.
There is currently no FDA approved treatment or cure for IPF and it is ultimately fatal. About two-thirds of patients diagnosed with IPF are no longer alive five years of the diagnosis.
It is important to determine the specific form of pulmonary fibrosis because the progression of the disease and the therapy used may differ depending on the cause. Learn more about ILD.
Only a subset of individuals with IPF have the familial variant of the disease.
Is FPF different than Idiopathic Interstitial Pneumonia?
Familial pulmonary fibrosis (FPF) is essentially the same disease as non-familial forms of IIP. The main difference between FPF and other non-familial forms of IIP is that there is a family history of IIP. This means there are at least 2 affected members in a family. Patients with FPF usually have similar symptoms, treatment options, and prognosis as individuals with non-familial forms of the disease. The onset of the disease is usually between the ages of 50-70 years.
 Clinical Trials
Clinical Trials
For more than 100 years, National Jewish Health has been committed to finding new treatments and cures for diseases. Search our clinical trials.